
New U Of I Research Points To Possible Cystic Fibrosis Treatment

The Illinois team specializes in molecular prosthetics, small molecules that could do the job of missing or deficient proteins. Pictured from left: Professor Martin Burke and graduate students Katrina Muraglia and Rajeev Chorghade. Fred Zwicky/Illinois News Bureau
Cystic fibrosis is a genetic disorder that causes people to suffer from frequent lung infections and difficulty breathing.
New research from the University of Illinois and University of Iowa points to a possible new treatment, a drug that the FDA has already approved to fight fungal infections.
Martin Burke—a chemistry professor at the University of Illinois and associate dean for research at the Carle Illinois College of Medicine—led the study.
Burke's research was published in the journal Nature Wednesday. He discussed it with Illinois Public Media’s Brian Moline.
This interview has been condensed and lightly edited for clarity
Brian Moline: I think most people have heard of cystic fibrosis but might not know that much about the disease? Could you give us a quick summary of cystic fibrosis?
Martin Burke: Sure. Many of us have in our lung a fully functioning protein called CFTR, which is a channel that allows bicarbonate, or baking soda, to be released from the surface of our lungs. This bicarbonate protects us against infections, and unfortunately in some people, this protein is missing or dysfunctional. And if you don't have the CFTR protein working properly, you can't release the bicarbonate, and therefore you are vulnerable to infections. People with CF (cystic fibrosis) have a loss of function of this protein and unfortunately end up with chronic lung infections, which are the primary driver of the disease. This is the challenge that we're trying to treat.
BM: Interestingly, the drug in this study is one that's been used to fight fungal infections and could help treat cystic fibrosis. How would it work?
MB: So, fundamentally, the problem in cystic fibrosis is a missing transporter function. And so what my lab discovered is that this small molecule, this medicine called amphotericin, forms channels in a way that can serve as a "molecular prosthetic" for the missing protein. So, just imagine: if you're missing a hand, of course, you can replace a missing hand with even a pretty simple prosthetic device that can actually restore a lot of function. This is really kind of the same thing happening on the molecular scale. So imagine like a molecular-scale prosthesis that is replacing the missing protein channel. It just happens to be a drug molecule that's acting as a surrogate.
As you point out, very fortunately, amphotericin is actually already a clinically approved drug that was known for very long time to form ion channels. And so we were able to test whether or not that channel-forming property could be used as a way to act as a molecular prosthetic for cystic fibrosis. And the reports in the study are, it's important to mention, all preclinical. So a lot of important work remains to be done to find out if this can be helpful for people with CF in the clinic. That all lies ahead of us. But the results that we've been able to generate thus far in the lab are very encouraging. And that's what we're excited to report in the study.
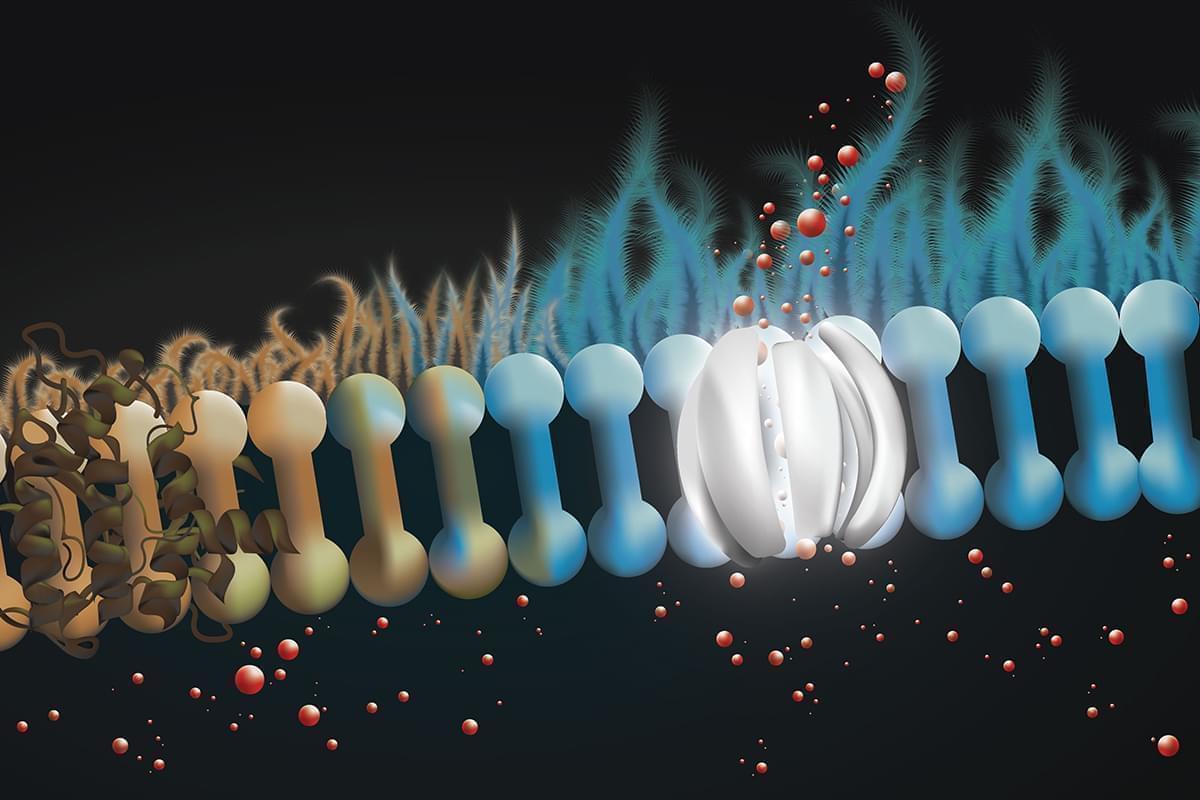
People with cystic fibrosis are missing a protein in the lining of the lung that releases bicarbonate, a key infection-fighting agent. The drug amphotericin can form channels to release bicarbonate in lung tissue, restoring the airway surface liquid’s antibiotic properties.
BM: You mentioned this drug amphotericin has this ability to form these channels. How long have you kind of known about this property of the drug? And then what was the process of deciding to give it a try in this type of research?
MB: The story goes back to when I was a graduate student in an MD-PhD program. So I was studying both chemistry and medicine at the same time. And when I was on my clinical rounds, I met an extraordinary young woman who had cystic fibrosis, and we were discussing in detail the deficiency of her protein function that was causing the problem. And in this conversation, at some point, she stopped me and said, 'It sounds like you know, exactly what's wrong, why can't you fix it?' And I'll tell you, it was really a life-changing conversation for me, because she had pointed out something, you know, really critical. And so there was this big gap between: we understood the problem, but there was very little we could do to actually address the problem.
I think it was those two experiences that really kind of crashed together in my mind that led to this idea."Dr. Martin Burke, University of Illinois
And at the same time, I was actually taking a chemistry class, and my professor introduced amphotericin as a very important problem in the field of synthetic organic chemistry, it had had a lot of work done on it. And he mentioned in that class that this molecule forms ion channels, and I think it was those two experiences that really kind of crashed together in my mind that led to this idea of: Could we use something like amphotericin as a channel-forming molecule to replace the missing protein channel in this young woman and other people with cystic fibrosis? So I actually applied for a job as an assistant professor here at Illinois, and was very fortunate to be given the opportunity to start my own lab really based on this idea. We proposed to try to go after this problem. We've been working on it for 14 years, and we're really excited to have made this important step forward. We're now in the lab, in cell culture, as well as in pigs, we're able to see that the amphotericin and channels look like they hold a lot of promise as a replacement for the CFTR.
BM: You mentioned the partnership with the University of Iowa on this project. How did that partnership come about?
MB: We've been very fortunate to work with Mike Welsh and his team at the University of Iowa. Mike is a world leader in cystic fibrosis. And my lab actually comes in as the outsider. So this wasn't what our lab was known for. This is our first paper, actually, on anything to do with cystic fibrosis specifically, and Mike's lab is a world expert. He has just tremendous depth of experience and instrumentation and knowledge about mechanism. And we actually formed a great strategic collaboration and partnership. And so we've benefited tremendously from this, my students have gone out and spent a lot of time in his labs at Iowa. Luckily, it's drivable. So they've been able to kind of just drive back and forth. And it's been a really great collaboration.
And we've benefited tremendously from working together to test this idea in some of the really advanced, hopefully, as clinically relevant model systems we could get our hands on. We have some people with CF that donate when they get a lung transplant. Mike Welsh is actually the developer of the best animal model for cystic fibrosis. He created a genetically modified pig that doesn't have the CFTR and it gets cystic fibrosis very much like people do. And so that was a really special opportunity to test the idea in a relevant animal model.
BM: How far along are you in this process? I guess I'll use a baseball analogy here. Let's say the top of the first inning would be the very start of the process. bottom of the ninth is getting this on the market as an approved treatment for cystic fibrosis. Where are you right now?
MB: I would say we're on the third or fourth inning. So we're not at zero, we're not just getting started. But we have a long way to go. It's really important to point out that we're very excited and optimistic about the results. But the next really important key step is going to be to find out if this can be helpful for people with CF in a clinical setting. So, a lot of road ahead of us, but a lot of optimism and hope based on what we've seen thus far.
We're not just getting started. But we have a long way to go."Dr. Martin Burke, University of Illinois
And one of the things that makes this approach really unique that I think we're particularly excited about is that because it doesn't actually depend on the CFTR protein itself. It could, in theory, work for everybody with cystic fibrosis, regardless of what mutation is causing you to lose the function and that distinguishes it from all the other approaches that are currently available in the clinic, which depend on a specific mutation of the protein. Whereas here, it's independent of CFTR, and therefore hopefully could be helpful to everyone. So this is why we're so excited, this might actually potentially add something really important to the clinical options available for patients.
BM: And what's the timeline on the clinical trials? When are you hoping to get those started?
MB: As soon as possible. We're committed to figuring out the safest and most effective and efficient way to answer this question in the clinic. And this is where we're putting all of our energy now, just figuring out what is the best path to actually do that.
BM: So of course, as you mentioned, still a long way to go. But it sounds like, Marty, you're pretty confident that this really has a lot of potential.
MB: I'd say we're hopeful. I'm hopeful. Confident is a strong word. Sure, it's challenging, right? I mean, obviously, it's a very complex problem and there's a lot of different angles to it, but I would certainly say we are hopeful and optimistic based on what we've seen thus far, and definitely committed to finding out if this could also be helpful for people with CF.
Links
- New Research Finds Microplastics In Illinois Groundwater
- Researchers To Explore Feasibility Of Industrial Hemp Industry In Illinois
- New U Of I Bioprocessing Research Lab Celebrates Grand Opening
- U of I Announces New Data Science Center, Research Park Expansion
- Researchers Engineer Bacteria To Create A Million New Chemicals For Drug Discovery
